
Por Carlos Campillo Artero, Servei de Salut de les Illes Balears
Tercer post de la serie de Evaluación Económica de Intervenciones Sanitarias (enlace al primero y al segundo). En el mismo Carlos Campillo revela un inquietante panorama del sistema de evaluación de aparatos y dispositivos sanitarios en Europa. En definitiva: mucho camino por recorrer.
Tan nocivo para la salud puede ser regular bajo captura como no regular por defecto o hacerlo con flacidez normativa y de control. Conocemos bastante el cuerpo regulatorio de los medicamentos, las consecuencias sociales de sus deficiencias y tenemos meridianamente claras las medidas que han de implantarse para solventarlas. Pero diversos estudios han señalado que los profesionales de la salud desconocen el funcionamiento de la regulación de los aparatos y dispositivos médicos (ADM).
En los últimos años se han puesto de manifiesto espacios de mejora de los mecanismos regulatorios de los ADM, sobre todo en los Estados Unidos y en Europa: eventos adversos asociados, por ejemplo, con prótesis de mama, prótesis de cadera y desfibriladores o la retirada del mercado por fallos de algunos ADM (aquí).
En los Estados Unidos, instituciones académicas y organismos oficiales como la General Accounting Office o el Institute of Medicine han difundido estudios e informes propios que ponen sobre el tapete hechos tales como que la eficacia y la seguridad de los ADM de alto riesgo sólo se han estimado mediante ensayos clínicos antes de la aprobación de su comercialización en el 27% de los casos, que un porcentaje creciente de ellos se aprueban a través de mecanismos regulatorios destinados a los de bajo riesgo, que ese porcentaje ha aumentado en los últimos años, y que las limitaciones de los ensayos que se realizan son graves. Cuando menos, conocen las características y la magnitud del problema.
En Europa, las investigaciones realizadas —si bien escasas y menos exhaustivas que las llevadas a cabo en Estados Unidos— apuntan en la misma dirección (aquí). Para entender cabalmente el problema y a modo de paréntesis debe señalarse que en Europa los ADM se clasifican en función del riesgo que suponen para la salud en: clase I (apenas acarrean riesgo alguno, por ej., estetoscopios, depresores linguales), IIa (su riesgo es moderado, por ej., ecógrafos, aparatos de resonancia magnética nuclear), IIb (su riesgo es algo mayor, por ej., aparatos que emiten rayos X), y III (mantienen vivo al paciente pero pueden poner en grave peligro su vida, por ej., desfibriladores implantables, stents, marcapasos, prótesis de cadera).
A diferencia de la regulación estadounidense (que centraliza la de medicamentos y ADM en la FDA), en Europa la de los medicamentos corre a cargo de la Agencia Europea del Medicamento, pero la de los ADM es responsabilidad de cada país miembro. Para poder comercializarse, los ADM deben satisfacer ciertos “requisitos esenciales”: pruebas de funcionamiento y fiabilidad acordes con sus riesgos inherentes (algo que decide el fabricante). Para conseguir comercializar los de clase I, el fabricante sólo debe presentar la documentación al regulador de su país. Además, los ADM han de recibir la autorización (con la marca CE: Comformité Européenne). Para esta clase la concede el propio fabricante. Los de clase IIa, IIb y III no son evaluados por la Agencia Europea, sino por uno de los 76 Notified Bodies (NB) acreditados por las autoridades competentes de cada país. Son empresas privadas, con fines de lucro, que cobran por evaluación y cuyo trabajo es financiado en parte por el fabricante, quien es libre de escoger el NB que le convenga. Es el NB quien concede la marca CE para estas clases de ADM. Al hacerlo, el ADM puede comercializarse en todos los Estados Miembros de la Unión Europea.
Los fabricantes de aparatos clases IIa, IIb y III han de presentar pruebas científicas de eficacia y seguridad de los ADM a un NB: pueden proceder de una revisión de la bibliografía, cuando el ADM es equivalente a otro comercializado, o de una revisión de ensayos clínicos, si persisten dudas sobre su seguridad. Para los de clase III, además, deben realizarse investigaciones clínicas con humanos (término ambiguo traducido al pie de la letra), aunque no necesariamente ensayos clínicos. Al no ser pública la información de este proceso, se desconoce su rigor y cumplimiento.
La normativa establece que estos requisitos se apliquen uniformemente en todos los países de la UE, pero cada NB aplica normas distintas y en conjunto utilizan métodos de evaluación heterogéneos. Cuando se deniega la comercialización de un ADM, se prohíbe tramitar una nueva solicitud por medio de otros NB, que no están obligados a notificar dichas solicitudes ni la concesión de la marca CE. La ausencia de registros centrales de solicitudes impide saber si estas se vuelven a presentar. Se desconoce qué ADM ha aprobado cada NB. Éstos no pueden actuar como consultores ni ayudar a los fabricantes a conseguir la autorización.
En Europa las evaluaciones se centran en el funcionamiento de los ADM más que en su eficacia clínica. Se carece de requisitos explícitos de eficacia absoluta (los de eficacia relativa o incremental son inexistentes) y no se exige certificar su seguridad sobre la base de pruebas clínicas. Las reformas del proceso regulatorio europeo no han ido a la par de los avances técnicos de los ADM, a despecho de las constantes modificaciones técnicas que se introducen en ellos.
La Unión Europea reconoce que la regulación vigente no puede sobrevivir sin introducir profundos cambios sistémicos, pero en varios análisis se vaticina que las reformas en curso no incidirán en la raíz del problema (aquí y aquí), a pesar de que se han difundido propuestas concretas de reformas, que incluyen medidas específicas (aquí y aquí).
En el caso de los ADM, las limitaciones de la valoración de su eficacia, seguridad y calidad y de su vigilancia postcomercialización son más marcadas que las de los medicamentos por los motivos señalados. Y la actividad del sector de los ADM en España no es escasa. Según la Memoria 2012 de la Federación Española de Empresas de Tecnología Sanitaria (aquí), si bien dicha actividad se redujo globalmente un 6%, se facturaron 7200 millones de euros, un descenso variable por servicios o productos (por ej., electromedicina (-45%), implantes (-8,6%), diagnóstico in vitro (-6%), cardiología (-5%), nefrología (-5,2%)). El valor de las exportaciones fue de 1.883 millones de euros (un 4,8% más que en 2011) y el de las importaciones, de 4360 millones (un 5% menos que en 2011).
En las decisiones de cobertura y fijación de precios de los ADM en España tampoco se aplican criterios de eficiencia. Esta omisión se produce, primero, aunque para los ADM tampoco exista motivo alguno para no hacerlo y no fijar precios en función de su coste-efectividad incremental. Y, segundo, a despecho de que la eficiencia conste en sucesivos reales decretos y órdenes como criterio para definir, detallar y actualizar la cartera de servicios comunes y de que se haya reconocido que la falta de énfasis en ella ha contribuido a conducir al SNS a una situación de grave dificultad económica (aquí).
Tampoco se conoce la influencia real que han ejercido en las decisiones de cobertura y fijación de precios las evaluaciones de ADM realizadas por las agencias de evaluación de tecnologías sanitarias españolas. Su cifra (Agencia de Evaluación de Tecnologías del Instituto de Salud Carlos III, Agencia de Calidad y Evaluación Sanitarias de Cataluña, OSTEBA (Agencia de Evaluación de Tecnologías del País Vasco), Agencia de Evaluación de Tecnologías Sanitarias de Andalucía, Servicio Canario de Planificación y Evaluación, AVALIA-T en Galicia y la recientemente extinguida Unidad de Evaluación de Tecnologías de la Agencia Laín Entralgo (Madrid)) y sus numerosos informes producidos se contraponen con la inacción regulatoria. No ha sido por falta de recursos.
En el acuerdo del Consejo Interterritorial del SNS, de 18 de marzo de 2010 se menciona que, además de considerar conveniente que la incorporación de nuevos medicamentos en la cartera de servicios del SNS ha de basarse en criterios de coste-efectividad, así como trabajar de manera conjunta para desarrollar guías farmacoterapéuticas que ayuden a que las decisiones clínicas se fundamenten en criterios de evidencia y coste-efectividad (tema abordado en este blog) se debería reforzar el papel de la Evaluación de Tecnologías Sanitarias. Se trabajará para reforzar las garantías y la seguridad en el procedimiento de autorización de las nuevas tecnologías en el SNS, mejorando la disponibilidad de evidencias científicas y de coste-efectividad como base para la toma de decisiones, mediante la creación de un modelo organizativo en red con las Agencias estatal y autonómicas.
En otro acuerdo de febrero de 2012 alcanzado por el Consejo Interterritorial del SNS se encomendó la creación de la Red de agencias de evaluación de tecnologías sanitarias y productos, encargada de generar información basada en la evidencia científica para la toma de decisiones el SNS. El RDL 16/2012 mantiene la función de esta red de ofrecer análisis de coste-efectividad para categorizar la cartera de servicios. No obstante estas normativas, la existencia de facto de trabajo en red entre agencias desde hace tiempo y de sus informes, de recursos de experiencias extranjeras, la evaluación económica, como ocurre con los medicamentos, también sigue sin aplicarse en España en las decisiones de cobertura y fijación de precios de los ADM. Cabe preguntarse cuánto tiempo se requiere para aplicar efectivamente los acuerdos realizados nada menos que en el marco del Consejo Interterritorial, máxima autoridad en materia sanitaria o para aplicar las leyes de carácter urgente, como consta en el RDL 16/2012.
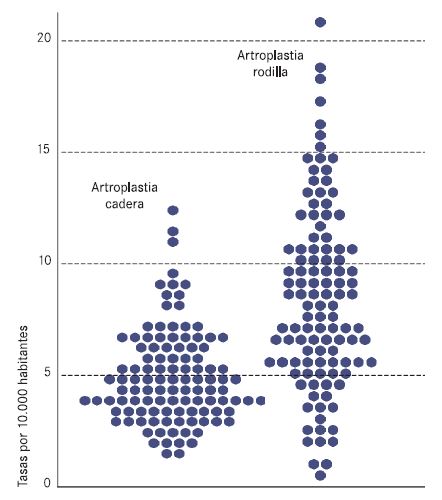
Un ejemplo meridanamente claro de la necesidad de incorporar la eficiencia en su regulación viene dado por la variabilidad injustificada de su uso, como ilustran los atlas de variaciones de las prótesis de rodilla y de cadera en España. La figura 1 (donde cada punto corresponde a un área de salud) muestra que las tasas estandarizadas por 10.000 habitantes en 2002 de artroplastia de cadera y rodilla (para tratar la artrosis) varían de 1,30 a 12,73 y de 0,97 a 20,58, respectivamente (véase aquí para otros ejemplos). Esta alta variabilidad se explica, fundamentalmente, por la discrecionalidad de los cirujanos de las distintas áreas de salud para indicar la intervención en situaciones de distinta gravedad (desde poco dolor y capacidad funcional hasta dolor muy intenso e imposibilidad de deambular).
Figura 1. Tasas de artroplastia de cadera y de rodilla por áreas de salud
El gasto evitable asociado con la utilización de ADM y la prescripción de medicamentos no justificadas clínicamente es elevado. Los recursos liberados podrían destinarse a financiar otras actuaciones con efectividad comprobada cuya financiación puede verse afectada por recortes indiscriminados.
Las causas del problema son multifactoriales. Por tanto, las soluciones han de ser multifacéticas, incidir sistémicamente en la regulación europea de los ADM y hacerlo con prontitud. La efectividad de algunos ADM está en juego, amén de la seguridad de algunos pacientes, como la confianza del público y la de los profesionales. Sin evaluación económica también los están la solvencia y sostenibilidad del SNS tal como aún lo disfrutamos hoy.

Hay 11 comentarios
Gracias por el post, Carlos. Dedicamos mucha atención a la regulación de los medicamentos que, con todos sus defectos, cuando menos asegura ciertos estándares de seguridad y eficacia (no siempre de efectividad y casi nunca de coste-efectividad), y no reparamos en el "blando" sistema regulatorio que se aplica a la comercialización de aparatos y dispositivos. Todavía me tiemblan las rodillas (y sin prótesis) pensando en el procedimiento de "evaluación" de los ADM con elección y financiación del organismo auditor por el propio frabricante-interesado.
Tienes toda la razón, Fernando. El del propio fabricante que comentas es lo de menos, porque se trata de ADM tipo I. Saludos,
Me parece increible ¿para cuando partidos políticos que pongan al consumidor en su frontispicio en vez de ser secuestrados por un sector empresarial?
Gracias por tu comentario, Estilpon.
Gracias Carlos por poner atencion a este tema tan dejado de la mano de los economistas, de la salud incluidos. Aunque no tuviera efecto alguno en la variedad de practicas observadas, no atender criterios de seguridad y coste efectividad ya hace merecedora la atencion. Buena entrada pues
Gracias por el comentario, Guillem. Tampoco olvidemos otro tema preterido de importancia creciente: las pruebas diagnósticas de laboratorio, tanto las nuevas (su valor diagnóstico incremental, su razón coste-efectividad incremental, incluidos nuevos biomarcadores cuya validación es limitada...), como las "small ticket" con altísima n incorrectamente solicitadas o las que deberían sustituirse por otras con mayor valor diagnóstico añadido (por ej., CK-MB-troponinas...). Algún que otro hospital en España a empezado a tomas medidas de desinversión y sustitución adecuadas, aunque las medidas son sólo incipientes.
Otra cuestion que no planteas pero que tiene su importancia. Los NB lo mismo te certifican un equipo de rayos X que un marcapasos implantable. Hasta ahora, esos NB sabian lo mismo de uno y de otro: practicamente nada. Los mas serios (TüV, SGS, etc) se estan poniendo las pilas y empiezan a aprender de que hablan, pero hasta no hace mucho era un autentico cachondeo, incluso cuando de verdad actuaban como entes independientes (y no como proveedores del fabricante, increible papel).
Sin contar con que la directiva 93/42 no es la unica que regula los dispositivos medicos, segun el tipo hay otras directivas que los rigen.
Y los marcados CE de algunos productos chinos o pakistanies, por poner solo dos ejemplos, son de risa.
Sin duda, elpep. Hay análisis de la "facilidad" con que el mismo producto recibe buenas valoraciones por parte de diferentes NBs. Esos dispositivos chinos, pakistaníes, coreanos y de otros países reciben marca como cuentas, al margen de los hallazgos del BMJ y The Daily Telegraph. Veremos cómo acaba la reforma anunciada.
Tampoco se puede esperar mucho de un pais sin una sola ingenieria en functional safety, como por ejemplo, la universidad de york . Los que nos dedicamos a safety somos autididactas .
Gracias por el comentario, Javier. ¿Por qué no nos explicas brevemente o nos das algunas referencia sobre functional safety y el papel que desempeñan los ingenieros? Gracias por adelantado.
Los comentarios están cerrados.