
 Me temo que este mes he sido poco original, y he decidido hablar de la secuenciación del ADN mitocondrial obtenido de los fosiles de la Sima de los Huesos en Atapuerca. Habrá leído usted sobre esto en infinidad de sitios, lo habrá visto en la prensa, y dirá: "Pero qué pesadez... ¿De verdad es tan importante esto?" Pues mire, sí, es muy importante, mucho; y a ver si soy capaz de explicarle por qué en lo que sigue.
Me temo que este mes he sido poco original, y he decidido hablar de la secuenciación del ADN mitocondrial obtenido de los fosiles de la Sima de los Huesos en Atapuerca. Habrá leído usted sobre esto en infinidad de sitios, lo habrá visto en la prensa, y dirá: "Pero qué pesadez... ¿De verdad es tan importante esto?" Pues mire, sí, es muy importante, mucho; y a ver si soy capaz de explicarle por qué en lo que sigue.
La pregunta ¿De dónde venimos? está muy lejos de contestarse. El linaje humano es una cosa muy complicada. Que si Homo erectus, que si Homo neanderthalensis, que si Homo sapiens... Que si nuestros antecesores, los Paranthropus o los Australopithecus... Una peña, vaya. Y nada bien organizada, además, repartidos por todo el mundo mundial, y encima en pequeños trozos, y tampoco muchos. Si se empeñan en ponerlo más difícil, no lo logran.
Visto desde fuera, da la impresión de que cada vez que un paleontólogo se tropieza con un hueso (o un molar, sin ir más lejos), ¡zas! Ya tenemos una nueva especie. Estoy simplificando, no es así; los paleontólogos son gente seria que usa criterios concretos para intentar discriminar lo qué es una nueva especie o lo que no. Pero un observador con el nivel PISA de nuestro estudiante medio podría creer que se asignan especies al buen tuntún. Claro, el problema casi que no es ese. El problema es que ahora que hemos desenterrado una costilla y tenemos al Homo montoriensis, ¿dónde lo colocamos en nuestra historia evolutiva?
Asignar su sitio evolutivo a H. montoriensis es una empresa complicada, y sujeta a discusión. A modo de ejemplo, consideremos un trabajo muy reciente en Science, en el que el equipo de David Lordkipanidze sugiere que muchas de las especies que se vienen reconociendo como tales son en realidad la misma; que las diferencias morfológicas entre los fósiles encontrados no son más que la variabilidad estándar en una especie. En apoyo de su propuesta, presentan resultados de cráneos encontrados en Dmanisi (Georgia) que están datados en fechas muy cercanas, recogidos en la misma zona, y son sin embargo muy diferentes. Así, no habría razones para distinguir entre H. habilis, H. rudolfensis, H. ergaster u H. erectus. Pero esta conclusión está muy lejos de ser unánimente aceptada, y como cuenta muy bien la investigadora del CENIEH María Martinón-Torres en el blog del Museo de la Evolución Humana, hay razones muy serias, basadas en análisis detallados, en particular de la dentición de los homínidos de Dmanisi, para pensar que estos georgianos primitivos podrían a su vez pertenecer a más de una especie, lo que invalidaría el razonamiento de Lordkipanidze y coautores. Y es que la identificación de especies por criterios morfológicos es siempre problemática, como también puede ver el lector en el post de Martinón-Torres.
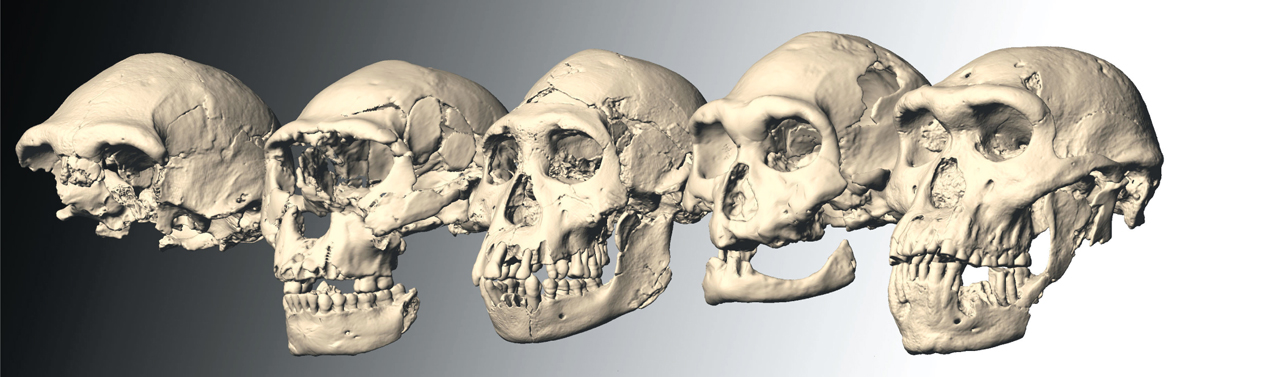
Figura 1. Los cráneos de Dmanisi; a la derecha, D4500, que fue portada del número de Science donde apareció el artículo.
Así las cosas, es necesario tener otras herramientas, otros criterios, para clasificar los fósiles que nos vamos encontrando, y poder así reconstruir la historia de nuestro linaje. Y es por eso por lo que el trabajo del ADN de los fósiles de la Sima de los Huesos es tan importante, porque es un salto cualitativo en ese trabajo de reconstrucción. Contra lo que sostienen algunos, basándose en criterios que no alcanzo a comprender muy bien, de que la matematización de las ciencias sociales las corrompe y las hace inútiles y de hecho perjudiciales, en mi opinión, y en consonancia con la filosofía de NeG de que es necesario apoyar las conclusiones en datos cuantitativos bien analizados, la técnica desarrollada por los autores del artículo nos va a permitir, andando el tiempo, aclarar nuestras ideas de manera definitiva.
Volvamos pues a la Sima de los Huesos. Hace unos 400.000 años, los cadáveres de al menos 28 individuos fueron arrojados, en algo que parece un enterramiento ritual, a un pozo en la Sierra de Atapuerca, dando lugar a lo que para nosotros es un yacimiento fósil único en el mundo. La presencia de tantos individuos permite sacar conclusiones bastante firmes sobre la especie a la que pertenecían estos homínidos, que resulta ser H. heidelbergensis. Este antepasado nuestro comparte, en su fisonomía, muchos rasgos con H. neanderthalensis, por lo que hasta ahora se viene considerando su antecesor directo. Para comprobar esta hipótesis, los autores del artículo procedieron a hacer un análisis de ADN de los restos fósiles. 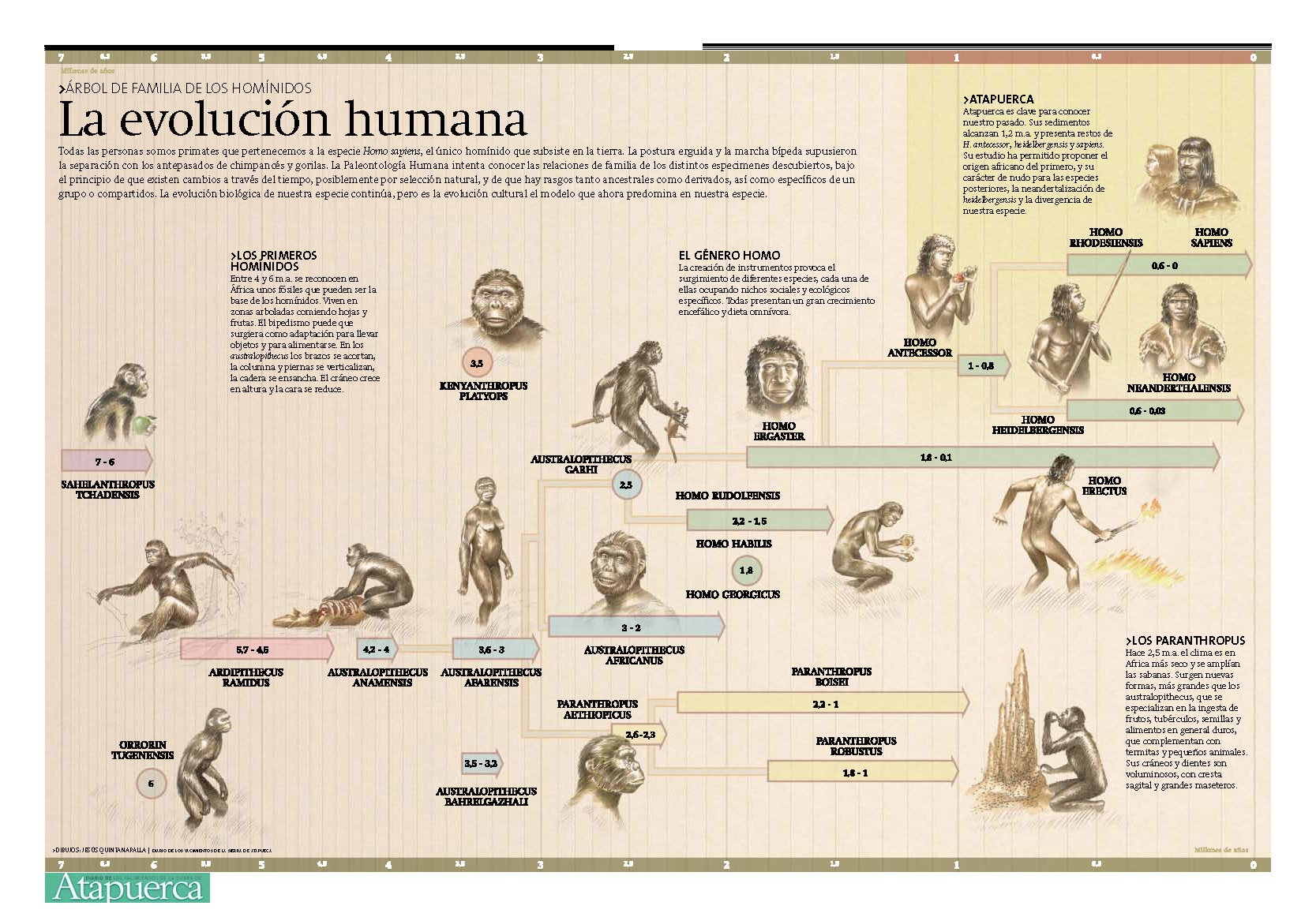
Figura 2. La evolución humana tal como se viene entendiendo, en un póster del Museo de la Evolución Humana de Burgos (original grande aquí).
Dicho así, parece todo muy fácil, pero analizar el ADN de fósiles de hace 400.000 años (del género Homo o del que sea, el problema es la edad) es enfrentarse a dificultades muy serias. La primera de ellas es, para empezar, extraer ese ADN. De hecho, el ADN al que se refiere el artículo no es el ADN nuclear, el que está presente en el núcleo de las células en forma de cromosomas, y que contiene la información genética para "construir" un ser humano. Tras trabajar con unos 2 g escasos (véase la foto de la entrada del post) de materia ósea de un fémur fosilizado, se logró extraer ADN mitocondrial, el contenido en un orgánulo de nuestras células llamado mitocondria y que proviene exclusivamente de la madre, ya que va incluido en el óvulo. Extraer ADN nuclear es todavía una asignatura pendiente; pero si tenemos ADN mitocondrial, ya podemos decir bastantes cosas. El siguiente paso es analizarlo. En este sentido, la propia antigüedad del ADN viene en nuestra ayuda; dado que la molécula se va deteriorando con el tiempo, en particular por sustitución de bases citosina por timina en los extremos de la molécula, se puede ver en cada trozo obtenido cuánto se ha degenerado y saber si es un trozo antiguo o nuevo. De esa forma, se pueden descartar todos los trozos que no corresponden al ADN del fósil; los que presentan pocas substituciones se asocian a contaminación por ADN humano moderno. Finalmente, hay que reconstruir la secuencia a partir de los trozos antiguos, es decir, "pegarlos" para producir el listado de las 16.000 y pico bases que conforman el ADN mitocondrial, teniendo en cuenta que esos trozos tienen esos cambios producidos por el tiempo, que provienen de distintas células que presentarían mutaciones y diferencias, más los trozos que directamente faltan o están poco representados... El proceso, análogo a reconstruir un libro partiendo de una colección de hojas sueltas y rotas de un grupo de copias de la obra, es laborioso y requiere de algoritmos sofisticados para el alineamiento de las secuencias. El resultado es que los investigadores pudieron secuenciar el genoma mitocondrial de manera casi completa.
Tras la carrera de obstáculos que acabo de describir, tenemos ya una secuencia de consenso (entre trozos, básicamente) del ADN mitocondrial del H. heidelbergensis. Ahora necesitamos más matemáticas; se trata de comparar esa secuencia con las de las demás especies para decidir cuál es el proceso evolutivo más probable que explique las diferencias observadas entre las secuencias de unas y otras. Esto es lo que se conoce como reconstruir el árbol filogenético que representa la historia evolutiva y el lugar de las especies consideradas en él, y que se hace mediante técnicas estadísticas con distintos programas que usan formalismos bayesianos. Uno de ellos, llamado Mr.Bayes, nos proporciona el árbol en sí; el otro, llamado BEAST, da el tiempo transcurrido desde el último ancestro común de cada dos especies. Para ello se utilizan, además del ADN encontrado en Atapuerca, los de humanos modernos, neanderthales, otros humanos conocidos como Denisovanos de los que luego hablaré, chimpancés y bonobos. El resultado de ese análisis es el que se recoge en la figura, que representa cuando fueron separándose unas especies de otras:
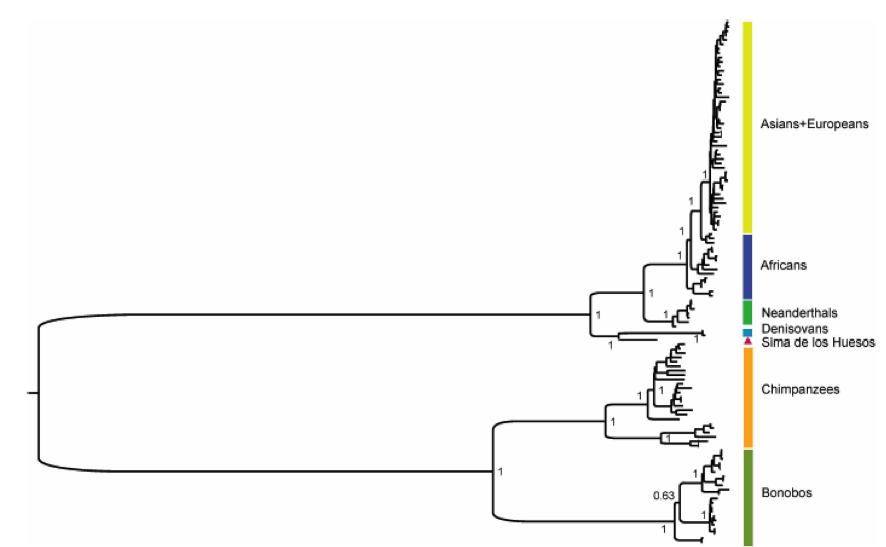
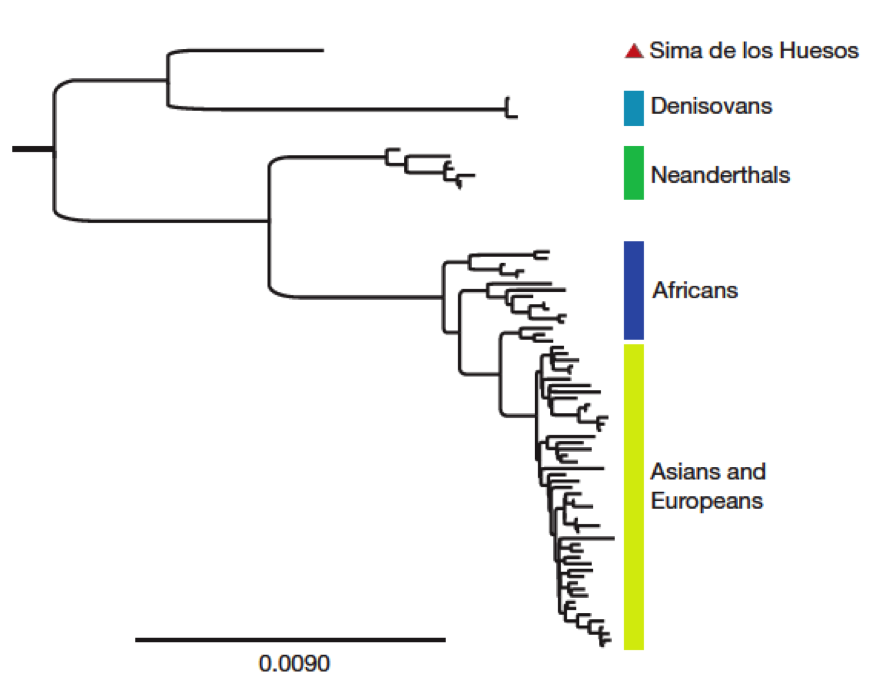
Figura 3. Árbol filogenético obtenido en la investigación a partir del ADN mitocondrial de la Sima de los Huesos. Arriba: árbol completo (pinchar para ver mejor). Abajo: Ampliación mostrando la separación de Neanderthales y hombres modernos por un lado y de los homínidos de la Sima y los Denisovanos por otro.
Y he aquí que, como dice la canción, "la vida te da sorpresas, sorpresas te da la vida". ¿Conque H. heidelbergensis era el "abuelo" del hombre de Neanderthal? ¡Ja! Como muestra la figura, el ADN mitocondrial del fémur de la Sima de los Huesos no está relacionado directamente con el Neanderthal, sino que ambos deberían proceder de una rama común que se bifurcó en épocas anteriores, dando lugar a dos ramas, una de las cuales originó luego a H. neanderthalensis y a nuestra propia especie, y la otra a H. heidelbergensis y... los Denisovanos, que son los más emparentados con los burgaleses de hace 400.000 años. Como dicen los autores del artículo, éste es un resultado totalmente inesperado. Los Denisovanos son unos recién llegados al listado de nuestros parientes, ya que fueron identificados en 2010 a partir de secuencias de ADN extraídos de una falange y un molar... ¡en Siberia! De ellos se sabe que están relacionados con los Neanderthales, pero de manera más lejana que nosotros mismos (como describe el arbol filogénetico de arriba), y poco más.
Así que ya tenemos un nuevo lío: ahora sabemos que H. heidelbergensis no es un antepasado del Neanderthal, y que está más cerca de los Denisovanos. ¿Pero cómo demonios unos fósiles de hace 400.000 años encontrados en Burgos van a estar emparentados con alguien que perdió una falange y un molar en Siberia, a nada, a unos cuántos miles de kilómetros? Pues la verdad es que los autores del trabajo no lo saben y dan hasta cuatro hipótesis para explicar sus resultados. De ellas, las que más probables les parecen son dos. En primer lugar, los homínidos de la Sima de los huesos podrían ser antepasados tanto de los Neanderthales como de los Denisovanos, aunque eso exigiría entender la posterior divergencia de estos dos grupos. La otra alternativa plausible es por intercambio genético entre la población de la Sima y otra población distinta que portara el ADN mitocondrial de los Denisovanos. Esto es, que distintos linajes humanos hubieran coexistido en el Pleistoceno medio. De hecho, hay fósiles encontrados en Europa que son más o menos coetáneos con los de la Sima pero que no tienen rasgos Neanderthal. Y también está la posibilidad de que ese cruce se realizara con descendientes del "patriarca" de Atapuerca, H. antecessor, que comparte rasgos con los H. erectus encontrados en Asia.
Como suele decirse, este trabajo plantea más preguntas de las que responde, pero las preguntas son sumamente interesantes porque nos muestran que todavía no sabemos nada de lo que ocurría con los humanos en la Europa de hace medio millón de años. Para continuar avanzando, habrá que obtener nuevas muestras de ADN mitocondrial de la Sima (no vaya a ser que justo hayamos dado con el homínido friki, y que no sea representativo) y, sobre todo, seguir trabajando para obtener ADN nuclear. Por otro lado, está claro que la paleontología y nuestra comprensión de la evolución humana sólo avanzará mediante el uso de técnicas cuantitativas como la de este caso. Hay otros ejemplos recientes de éxitos en este sentido: Así, un estudio comparativo de 1200 molares y premolares de 13 especies homínidos ha mostrado que ninguno de ellos es el antecesor común de los humanos modernos y los Neanderthales, y que ambos linajes podrían haber divergido hace un millón de años. Otro trabajo cuantitativo muestra que el análisis de los patrones de corte en los huesos de animales consumidos por los homínidos (y su comparación con patrones obtenidos por experimentos llevados a cabo hoy en día) permite identificar la estandardización de los procesos de extracción de la carne y obtener conclusiones sobre su transmisión por aprendizaje y las distintas ocupaciones de un mismo yacimiento. En definitiva, estamos ante una nueva era en la investigación en evolución humana, al estilo Nada es Gratis.
Actualización de última hora: ayer se publicó en Nature un nuevo trabajo del equipo de Svante Pääbo, coautor del aquí reseñado, con el genoma completo de una mujer Neanderthal de hace unos 50.000 años. Las conclusiones son que en su pasado había un alto grado de parentesco entre sus antepasados, y que hubo flujo de genes entre Neanderthales, Denisovanos y humanos modernos, además de quizá un cuarto grupo más antiguo. Esto se pone cada vez más interesante...
P.S. Randy Schekman, que recogió su premio Nobel de Química esta semana, alertó esta semana en una carta a The Guardian sobre la desmedida influencia de revistas como Nature, Science o Cell en la ciencia, al extremo de llegar a pedir un boicot (aquí la noticia en castellano). ¡Más de lo que puede aguantar mi ego! ¿Cómo voy a dejar pasar la oportunidad de decir "ya lo dije yo en este foro"? Pues dicho queda: ya lo dije yo en este foro (aunque no llamé al boicot; lo aclaro no sea que luego me boicoteen a mí...).
Hay 6 comentarios
me apasiona este tema. Hace poco vi por algun lado que H. Georgensis podría descender de una población de Habilis, y que H. Floresiensis tiene un rasgo muy arcaico en los huesos de la muñeca, creo recordar, que le emparentarían más con los dos anteriores que con Erectus. Menudo puzle
saludos
Anxo hace muy bien en pegarle palos al bruto de E.O. Wilson: http://online.wsj.com/news/articles/SB10001424127887323611604578398943650327184
Gracias JR76. Vi ese artículo cuando salió y me indignó, la verdad. Sobre todo porque Wilson siempre me ha parecido un gran científico, y ha hecho aportaciones de gran calado. Pero creo que (quizá debido a su avanzada edad) está acabando su carrera 'molt malament'.
Bueno, algo de razón tiene Wilson. Una cosa es la matemática pura (una diciplina maravillosa, por cierto) y otra la matemática aplicada. El uso de la segunda en las ciencias factuales siempre debe estar supeditado a unos métodos y objetivos generales que no son de índole matemática. Por otra parte, cada campo concreto de las ciencias factuales requiere normalmente recurrir a un campo concreto, y sólo a ese, de la matemática. Por ejemplo, en genética parece claro que el uso de la estadística se ha vuelto imprescindible, pero dudo que un buen biólogo especializado en ese campo tenga que conocer, por ejemplo, la geometría diferencial.
En economía, precisamente, la hipermatematización es una de las causas de la actual decadencia de esta disciplina.
Una puntualización: Es bastante hipócrita hablar de la excesiva influencia de revistas como Science ó Nature cuando uno fue parte integrante de su sistema de publicación beneficiándose del mismo y el personaje en cuestión está interesado en que una nueva revista como eLife alcance el mismo interés mediatico. Si envías un artículo a Nature, Science o Cell y uno a eLife donde Randy es editor y comparas las respuestas al rechazarlo ves que es más de lo mismo.
Los comentarios están cerrados.